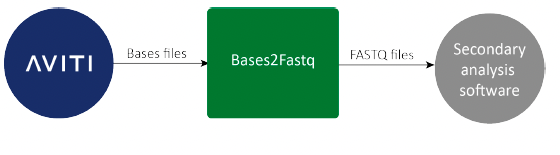
Bases2Fastq
The Bases2Fastq Software demultiplexess sequencing data and converts base calls into FASTQ files for secondary analysis with the FASTQ-compatible software of your choice. The Element AVITI™ System records base calls, which are the main output of a sequencing run, with associated quality scores (Q-scores) in bases files. Bases files must be converted into the FASTQ file format for secondary analysis.
This workflow guide provides informationon running Bases2Fastq, including system requirements,commands and arguments,a nd input and output files. Use is subject to the license available at go.elembio.link/eula.
Figure 1: Converting bases into FASTQ files
Software Features
Bases2Fastq runs off-instrument and has a command-line interface (CLI). A generalrun commandexecutesthe software.A Bases2Fastqrun startswithdemultiplexing,whichidentifieseach sample by the index sequence and assigns polonies based on the sequence. If samples are not indexed, Bases2Fastq skips demultiplexing and assigns all polonies to one sample. Bases2Fastq converts the demultiplexed bases into FASTQ files, generating one FASTQ file per read (e.g.,Read 1 or Read 2) per sample.
Arguments appended to the general run command let you adjust demultiplexing and FASTQ file generation to suit the application and leverage the following features.
For a complete list of options, see Optional Arguments.
- Paired-end and single-end adapter trimming, including detection of adapter sequences
- Base masking that includes or excludes cycles from FASTQ files
- Index sequence orientation detection,which you can enable or disable
- Quality control (QC) reports that open in a browser
- Unique molecular identifier (UMI) support
Run Manifest
A run manifest is a comma-separatedvalues (CSV) file that specifies demultiplexing settings, FASTQ file settings,and sample information. Bases2Fastq uses the run manifest to execute a run, by default accessing the output run manifest named RunManifest.csv in the run folder.
Run Bases2Fastq with the output run manifest or a prepared corrected version. You can also use Bases2Fastq arguments to override run manifest settings.The Run ManifestWorkflowGuide(MA-00011) provides complete information, including preparation instructions.
Default Run Manifest
If you do not upload a prepared run manifest during run setup, the AVITI System creates a default run manifest based on the run parameters entered. A default run manifest does not contain index sequences and assigns all reads to one sample during FASTQ generation.
Demultiplexing indexed librariesis not possible with a defaultrun manifest. If sequencing indexed libraries, prepareand uploada run manifest with index sequencesto the AVITISystem. Alternatively,you can create a corrected run manifest with all samples and their associated index sequencesto use with Bases2Fastqafter sequencingcompletes.
Corrected Run Manifest
To create a corrected run manifest,updatethe run manifestused for the sequencingrun. Run Bases2Fastq with the corrected version. For an example,see Run a CorrectedRun Manifeston page 18
The followin gcases requirea corrected run manifest:
- You sequencedindexedlibrarieswitha run manifestthat does not includeindexsequences.
- A run manifest failed or reported incorrect resultsdue to incorrect settings or indexes,requiringreprocessing.
- During sequencing,you did not upload a run manifest with indexed samples. Use a corrected run manifest with all samples and their associated index sequences to demultiplex.
Software and System Setup
Bases2Fastq is run as eithera staticbinaryexecutableor in a containerizedexecutionwithDocker.Therefore,settingup Bases2Fastq requires setting up Docker or the static binary on a computer that meets systemrequirements.The computermust also be configured to transfer input and outputfiles.
Bases2Fastq cannot run on an arm processor.
System Requirements
The computer running Bases2Fastq must meet the following requirements:
- Operating system (OS)—Docker runs on any OS. Static binary requires LinuxOS on an x86 architecture with glibc 2.17 or later installed. To verify the glibc version for static binary,run
ldd --version. - Memory—Both Docker and static binary require 4 GB RAM per concurrent thread.
Software performance depends on the resources dedicated to the processing environment. Element recommends running Bases2Fastq in the cloud on the Amazon m5dn.12xlarge EC2 instance,a virtual server,for optimal performance.Review instance specifications at aws.amazon.com/ec2/instance-types/m5/.
File Transfer and Storage
Store input and output files in a local location or the cloud using Amazon Web Services (AWS), Google Cloud Storage (GCS), or an rclone-compatible provider. Transferring files requires paths to the input and output locations. If the locations are both AWS or both GCS, they must use the same set of credentials.
When using cloud storage,Bases2Fastq downloads input files and stages output files in a temporary directory. The temporary directory must have ~800 GB of scratch space. By default, Bases2Fastq uses the temporary directory of the OS. To change the location of the temporary directory,set the environment variable TMPDIR. In Linux,run the command export TMPDIR="/path/to/scratch", replacing /path/to/scratch with the desired directory.
AWS Storage
AWS storage requires Uniform Resource Identifiers (URIs) to serve as pathsto the Amazon Simple Storage Service(S3) buckets that containthe inputand outputfiles. Accessing the buckets requires AWSc redentials configuredf or the setup.
- For AWS withan EC2 instance, Bases2Fastq detects the credentialsor Identity and Access Management (IAM) role associated with the instance.
- For AWS without an EC2 instance,define AWS credentials with the following environment variables:
- AWS_ACCESS_KEY_ID
- AWS_SECRET_ACCESS_KEY
- AWS_DEFAULT_REGION
GCS Storage
GCS storage requires Uniform Resource Locators (URLs) to serve as preconfigured paths to the Cloud Storage buckets that contain the input and output files. Set the environmentvariableGOOGLE_APPLICATION_CREDENTIALS to link to the file containing applicationcredentials.
Rclone-Compatible Storage
The file transfer service allows Bases2Fastq to access cloud storage locations.The service is compatible with many cloud storage providers. owever,Element has not tested every available rclone provider.
Followthe instructions at rclone.org/install to download and install rclone. Configure rclone to communicate with your cloud storage per the storage provider-specific instructionsat rclone.org/#providers.
Docker Setup
To downloadand install Docker, follow the OS-specific instructions at docs.docker.com/get-docker/. Element maintains a public registry at hub.docker.com/repository/docker/elembio/bases2fastq.
Static Binary Setup
Static binary setup requires downloading and extracting the static binary. Current and previou sversions are available for installation.
Set Up the Current Version of Bases2Fastq
- Download the static binary using one of the following methods:
- Visit go.elementbiosciences.com/bases2fastq-download and follow the onscreen prompts.
- Run the wget command
wget https://bases2fastq-release.s3.amazonaws.com/bases2fastq- latest.tar.gz.
- Run the curl command
curl https://bases2fastq-release.s3.amazonaws.com/bases2fastq- latest.tar.gz.
- Runtar -xzvf bases2fastq.tar.gz to extractthe downloadedfile.
- Run
./bases2fastq --versionto displaythe software version and confirm that Bases2Fastq is operational. - To generate HTML QC reports,run the following commands to install Python 3.6 or newer with NumPy,Bokeh,and bs4 packages:
sudo apt install python3 python3-pip libjpeg-dev zlib1g-dev
pip3 install numpy==1.\* bokeh==2.\* bs4==0.\*
—The bs4 package requires Pillow, which in turn requireslibjpeg-dev andzlib-dev. If you do not install Python 3.6 or newer or a package is missing, Bases2Fastq logs a warning and does not generate the report.—
Set Up a Previous Version of Bases2Fastq
- Visitgo.elembio.link/documentationto reviewreleasenotes for previousversionsof Bases2Fastq.
- Download the static binary using one of the following methods:
- Run the following wget command with a version number in place of
<version>:
wget https://bases2fastq- release.s3.amazonaws.com/bases2fastq-<version>.tar.gz
- Run the following curl command with a version number in place of
<version>:
curl https://bases2fastq- release.s3.amazonaws.com/bases2fastq-<version>.tar.gz.
—For example, to use wget to download Bases2Fastq v1.2.0, run the commandwget https://bases2fastq- release.s3.amazonaws.com/bases2fastq-v1.2.0.tar.gz.—
- Run
tar -xzvf bases2fastq.tar.gzto extract the downloaded file. - Run
./bases2fastq --versionto display the software version and confirm that Bases2Fastq is operational. - To generate HTML QC reports,run the following commandsto install Python 3.6 or newer with NumPy ,Bokeh, and bs4 packages:
sudo apt install python3 python3-pip libjpeg-dev zlib1g-dev
pip3 install numpy==1.\* bokeh==2.\* bs4==0.\*
—The bs4 package requires Pillow, which in turn requireslibjpeg-dev andzlib-dev. If you do not install Python 3.6 or newer or a package is missing, Bases2Fastq logs a warning and does not generate the report.—
Running Bases2Fastq
- Type the general run command that applies to your storage solution.
| Storage | General Run Command |
|---|---|
| AmazonS3 | bases2fastq s3://bucket/input s3://bucket/output [options] |
| GCS | bases2fastq gs://bucket/input gs://bucket/output [options] |
| Local | bases2fastq /path/to/input /path/to/output [options] |
| rclone | bases2fastq path/to/input/remote path/to/output/remote --input-remote "input-name" --output-remote "output-name" [options] |
—When using Docker, the general run command automatically pulls the image to your local environment.—
- Replace the input and output locations in the general run command.
| Storage | Location to Replace | Replacement |
|---|---|---|
| AmazonS3 | s3://bucket/input | URI to the AmazonS3 bucket that stores the input files |
| s3://bucket/output | URI to the AmazonS3 bucketthat stores the FASTQ files and other outputs | |
| GCS | gs://bucket/input | URI to the Cloud Storage bucket that stores the input files |
| gs://bucket/output | URI to the GCS bucket that stores the FASTQ files and other outputs | |
| Local | /path/to/input | Directory where Bases2Fastq accesses the input files |
| /path/to/output | Directory where Bases2Fastq writes the FASTQ files and other outputs | |
| rclone | path/to/input/remote | Provider path to the rclone remote that points to the input files |
| input-name | Name of the rclone remote that pointsto the input files | |
| path/to/output/remote | Provider path to the rclone remote that points to the FASTQ files and other outputs | |
| output-name | Name of the rclone remote that points to the location of the FASTQ files and other outputs |
- Adjust arguments in the general run command.
- To add arguments,replace
[options]with any of the arguments listed in OptionalArguments - To run Bases2Fastq without any arguments, delete
[options].
- To add arguments,replace
- Press Enter to run Bases2Fastq. —The terminal displays run progress.—
- Wait for the terminal to display the elapsed time, which indicates that processing is complete.
- Access the output files in the location specified in the general run command.
- To viewlogs, go to
info/Bases2Fastq.log. - To view the HTML QC report, go to the output folder, double-click the file, and move through each tab.
- To viewlogs, go to
Optional Arguments
The following arguments are optional additions to the general run command. Arguments that affect run parameters default to a value recorded in RunParameters.json, which is an output of a sequencing run and an input for Bases2Fastq.
| Argument | Command | Default |
|---|---|---|
| --chemistry-version | Overwrite the sequencing kit version. Valid values are 1 for a version 1 kit or Cloudbreak for a Cloudbreak™ kit. | RunParameters.json |
| --demux-only,-d | Enable demultiplexing-only mode, which performs demultiplexing and outputs index metrics without generating any FASTQ files. | False |
| --detect-adapters | Detects adapters sequences, overriding any sequences present in the run manifest. | False |
| --exclude-tile,-e | Use regular expression (regex) to specify a subset of tiles to exclude from processing(e.g.,L1.*C02S.). To specify multiple subsets, enter the argument multiple times. | Not applicable |
| --flowcell-id | Overwrite the flowcell ID used for a run. | RunParameters.json |
| --force-index-orientation | Perform demultiplexing withoutd etecting the index sequence orientation. When true, Bases2Fastq applies the orientation recorded in the run manifest. | False |
| --help, -h | Display the usage statement. | Not applicable |
| --i1-cycles | Overwrite the number of cycles in Index1. | RunParameters.json |
| --i2-cycles | Overwrite the number of cycles in Index2. | RunParameters.json |
| --include-tile,-i | Use regex to specify a subset of tiles for processing after excluding all tiles from processing(e.g.,--exclude-tile ".\*" --include- tile "L1.\*C02S."). To specify multiple subsets, enter the argument multiple times. | Not applicable |
| --input-remote | Sets the rclone remote name that points to the input files. | Not applicable |
| --kit-configuration | Overwrite the kit configuration. Valid values are 150Cycles for a 2 x 75 kit or 300Cycles for a 2 x 150 kit. | RunParameters.json |
| --legacy-fastq | Applya legacy file naming convention to FASTQ files, such as SampleName_S1_L001_R1_001.fastq.gz. | False |
| --log-level, -1 | Specify the minimum level required to log an event:INFO, DEBUG,WARNING,or ERROR. | INFO |
| --num-threads,-p | Specify the number of threads to use for processing.The minimum value is 1, and the maximum value depends on your system. | 1 |
| --num-unassigned | Specify a value ≤ 1000 that indicates the maximum number of unassigned sequences to support. | 30 |
| --output-remote | Sets the rclone remote name that points to the location of the FASTQ files and other outputs. | Not applicable |
| --preparation-workflow | Overwrite the library prep workflow. Valid values are Adept and Elevate. | RunParameters.json |
| --qc-only | Enable QC-only mode, which generates a representative view of run metrics on one tile without generating any FASTQ files. | False |
| --r1-cycles | Overwrite the number of cycles in Read 1. | RunParameters.json |
| --r2-cycles | Overwrite the number of cycles in Read 2. | RunParameters.json |
| --run-manifest,-r | Overwrite the location of RunManifest.csv, which is the run manifest the run generated, with the path to another run manifest. | Not applicable |
| --settings | Overwrite run manifest settings by entering the argument for each setting you want to overwrite. For example: - —settings "I1Fastq,True” - —settings "I2Fastq,True”- settings "I1Mask,I1:N3Y* | Not applicable |
| --skip-qc-report | Do not create an HTML QC report. | False |
| --split-lanes | Divide FASTQ files by flowcell lane. | False |
| --strict,-s | Enable strict mode, which prevents processing when any input files are invalid. When strict mode is off, Bases2Fastq warns you that files are missing or corrupt but continues processing. | False |
| --version,-v | Display the current version of Bases2Fastq. Bases2Fastq logs the version at the start of FASTQ file generation regardless of whether you include this argument. | Not applicable |
- The available setting sare listed in Run ManifestSettingson page 13
Additional Commands
Bases2Fastq supports the help and version argumentsas standalone commands that you can run without the general run command.
I did not include the run manifest settings as it can be linked.
Adapter Trimming
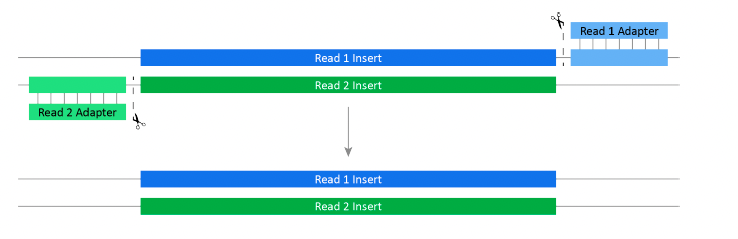
Library prep adds Read 1 and Read 2 adapters to each sample. When the length of Read 1 or Read 2 exceeds the length of the DNA insert, the run sequences into the adapter.Adapter trimming removes the adapter sequences from the 3′ end of each read to prevent adapter-based errors in certain analyses.
Run manifest settings enable adapter trimming and specify the options. When adapter trimming is enabled, Bases2Fastq automatically detects and trims adapter sequences regardless of whetherthe run manifest specifies the adapter sequences.
Figure 2: Trimming adapter sequences from Read 1 and Read 2
Paired-End versus Single-End
Bases2Fastq includes paired-end and single-end adapter trimming.Paired-end adapter trimming aligns the Read 1 and Read 2 inserts to accurately trim short adapters. When a sample includes insertions and deletions (indels), the software accurately trims adapters that are as short as one base. Single-end adapter trimming individually processes each read, removing the adapter sequences without alignment.
Paired-end adapter trimming is more accuratebut requires that Read 1 and Read 2 each include at least 17 cycles. Single-end adapter trimming supports applications hat do not meet this requirement. Neither type of adapter trimming increases the run time.
Default Adapter Sequences
The default R1Adapter and R2Adapter valuesfor the Adept Workflow are blank. Consult the third-party library prep documentation for adapter trimming recommendations. If you do not specify values, Read 1 and Read 2 must each include at least 48 cycles. Otherwise, Bases2Fastq cannot detect and trim the adapters.
For the ElevateWorkflow, the following sequences are the default values:
- R1Adapter—5' ATGTCGGAAGGTGTGCAGGCTACCGCTTGTCAACT 3'
- R2Adapter—5' ATGTCGGAAGGTGTCTGGTGAGCCAATCCAGCACG 3'
Base Masks
A base masks pecifies a set of cycles for a demultiplexing operation. Within a base mask,a series of operators indicates whether cycles are masked. A positive integer or asterisk follows each operator to indicate which cycles to mask.
- A Y (yes) operator indicates that a cycle is included in the mask.
- An N (no) operator indicates that a cycle is excluded from the mask.
- A positive integer indicates the numberof cycles to include or exclude.
- An asterisk matches any remaining cycles in the read.
For example,Y4N* masks the first four cycles in a read.The base mask N3Y2N* leaves the first three cycles of a read unmasked, masks the fourth and fifth cycles,and leaves the remaining cycles unmasked.
Read Identifiers
A base mask can include read identifiers that restrict the mask to Index1, Index2, Read 1, or Read 2. Each read identifier is encoded as the abbreviated read name followed by a colon (e.g.,R1:). If the base mask does not include a read identifier,Bases2Fastq uses a default read that depends on the following settings: I1Mask, I2Mask, R1FastQMask, R2FastQMask, and UmiMask.
To specify one read for a base mask, start the base mask with the read identifier. If you are specifying multiple reads for a base mask, enter multiple read sections that each start with the read identifier. Separate each read section with a plus sign.
- Example base mask that applies to one read:
I1:Y3N* - Example base mask that applies to two reads:
I1:Y3N*+I2:Y2N*
Cycle Lengths
A base mask must define the full cycle length of a read, regardless of whether you mask select bases in the read or all bases. A read with select bases masked must still account for the remaining cycles. Otherwise,Bases2Fastq displays a validation error.
For example, if Read 1 consists of 30 bases and you want to mask the first 15, end the base mask with the remaining number of cycles. The base mask R1:Y15N15masksthe first 15 bases (Y15) of Read 1 (R1:) and leaves the remaining 15 bases unmasked(N15). Alternatively, R1:Y15N* achieves the same goal but uses an asterisk to cover the remaining number of cycles.
Example Base Masks
| Base Mask | Result | |
|---|---|---|
R1:Y2N* | Matchesthe firsttwo cycles of Read 1 | |
N3Y2N3 | Matchesthe fourthand fifthcycles of the defaultread !(Aspose.Words.dda2bdc7-cf9e-40a0-a542-84989225fc34.028.png) | |
I1:N2Y*N2 | Matches all but the first two and last two cycles of Index1 | |
R1:Y*N-R2:Y*N | Matches all but the last cycles of Read 1 and Read 2 |
Example Run Commands
The following sections provide example run commands, which demonstrate how to perform the following functions:
- Run Bases2Fastq in QC-only mode.
- Run Bases2Fastq with a corrected run manifest. For more information,see Corrected Run Manifest
- Run Bases2Fastq with the settings argument to adjust run manifest settings. For a list of settings,see Run ManifestSettings* on page 13
Run QC-Only Mode
Static Binary
./bases2fastq /path/to/input /path/to/output --qc-only!(Aspose.Words.dda2bdc7-cf9e-40a0-a542-84989225fc34.034.png)
Docker
docker run --rm -v </path/to/input>:/input -v </path/to/output>: /input /output --qc-only
Run a Corrected Run Manifest
Static Binary
./bases2fastq /path/to/input /path/to/output -r /path/to/corrected\_ manifest\_filename.csv!(Aspose.Words.dda2bdc7-cf9e-40a0-a542-84989225fc34.036.png)
Docker
docker run --rm -v </path/to/input>:/input -v </path/to/output>:/output bases2fastq!(Aspose.Words.dda2bdc7-cf9e-40a0-a542-84989225fc34.036.png) /input /output -r /input/<corrected\_manifest\_filename.csv>
Adjust Run Manifest Settings
Static Binary
- The following example generates a FASTQfile for Index1 and a UMI FASTQ file.
./bases2fastq /path/to/input /path/to/output --settings "I1Fastq,True" --settings!(Aspose.Words.dda2bdc7-cf9e-40a0-a542-84989225fc34.037.png) "UmiFastQ,True"
- The followingexampleenablessingle-end adaptertrimmingfor Read 2.
./bases2fastq /path/to/input /path/to/output --settings "R2AdapterTrim,True"!(Aspose.Words.dda2bdc7-cf9e-40a0-a542-84989225fc34.037.png) --settings "AdapterTrimMode,Single-End"
Docker
- The followingexamplegeneratesa FASTQfile for Index1 and a UMI FASTQ file.
docker run --rm -v </path/to/input>:/input -v </path/to/output>:/output!(Aspose.Words.dda2bdc7-cf9e-40a0-a542-84989225fc34.038.png) bases2fastq /input /output --settings "I1Fastq,True" --settings "UmiFastQ,True"
- The following example enables single-end adapter trimmingfor Read 2.
docker run --rm -v </path/to/input>:/input -v </path/to/output>:/output!(Aspose.Words.dda2bdc7-cf9e-40a0-a542-84989225fc34.038.png) bases2fastq /input /output --settings "R2AdapterTrim,True" --settings "AdapterTrimMode,Single-End"
Inputs and Outputs
Inputs to Bases2Fastq are the files that a sequencing run generates and transfers to a run folder in your storage location. In turn, Bases2Fastq generates an output folder that containsthe outputfiles,an info directory,and a Samplesdirectory.The Samples directoryorganizesFASTQ filesand samplemetricsinto one folderfor each sample.
Figure 3: Bases2Fastq output directory
Input Files
The followingtableliststhe filesthat serve as inputfor Bases2Fastq. Bases2Fastqonlyuses the alignmentfile whenthe run manifest settingSpikeInAsUnassignedis set to true.
| File | Directory and File Name | Description | Quantity* |
|---|---|---|---|
| Alignment | Root/Alignment/{read}_{tile}.aln | Binaryfilesthat indicatewhichpolonies alignto PhiXControlLibrary | One per tile per read |
| Bases | Root/BaseCalls/{tile}/{read}_ {tileName}_C{cycle:000}.bases.gz | Binaryfilesthat containbase calls and associatedQ-scores | One per read, tile, and cycle |
| Filter | Root/Filter/{tile}.filter | Binaryfilesthat containthe filterstatus for each polony | One per tile |
| Location | Root/Location/{tile}.loc | Binaryfilesthat identifypolonylocations on the flowcell | One per tile |
| Run manifest | Root/RunManifest.csv CSV file that records biologicalsample informationand analysissettings | One per run | |
| Run manifest | Root/RunManifest.json JavaScriptObjectNotation(JSON)file reservedfor Elementprocesses | One per run | |
| Run parameters | Root/RunParameters.json JSON filethat records information aboutthe run configuration | One per run |
Output Files
The followingtableliststhe filesthat Bases2Fastqoutputs.Subsequentsectionsdetailthe HTML QC report,FASTQfiles,run metrics, and sample metrics.
| File | Directory | Description |
|---|---|---|
| Bases2Fastq.log | info | Log file that records softwareevents |
| HTML QC report | Root | Interactive report on run performanceand quality! |
| FASTQ | Samples/{sample} | The primary output of Bases2Fastq |
| IndexAssignment.csv | Root | Yield and the number and rate of polonies assigned for each sample and index combination |
| Metrics.csv | Root | The mismatchrates, percent assigned,and per-sample yield or each lane |
| RunManifest.csv | Root | The AVITI Operating Software (AVITIOS)- or user-created run manifest |
| RunManifestErrors.json | info | A record of errors in the run manifest |
| RunParameters.json | Root | A copy of the originalrun parametersfile |
| RunStats.json | Root | Informationon run performance |
| Samplemetrics | Samples/{sample} | Information on the performance of each sample in the run |
| UnassignedSequences.csv | Root | The most frequent unassigned index sequences with approximate counts |
HTML QC Report
The HTML QC reportopens in a browserso you can move throughvarioustabs.The tabs let you reviewhistogramsand other charts that visualizeindexassignmentand other qualitymetrics.Bases2Fastqnames the reportper the convention{runname}_QC.html. The reportdoes not showper samplecharts if more than 96 samplesare presentin the run manifest.
FASTQ Files
A FASTQfile records all genomic data and corresponding Q-scores for a sample.FASTQfilesare GZIP-compressedtextfilesnamedper the convention{sample}_{read}.fastq.gz.
Each entryin a FASTQ file corresponds to one read and includes the following four lines:
- A sequence identifier that includes run and polony information
- Base calls assembled into a sequence comprised of A, C, G, T, and N
- A plus sign (+) that separates the sequencefrom the Q-scores
- A Q-score for each base in the sequence
Sequence Identifiers
A sequence identifier includes the components described in the following table, formatted in one line: @<instrument>:<run name>:<flow cell ID>:<lane>:<tile>:<x-pos>:<y-pos>:UMI <read>:N:0:<index sequence>.
| Component | Value | Description |
|---|---|---|
| @ | @ | Start to the sequence identifier line |
<instrument> | Upper and lowercase letters, integers 0-9, and underscores (_) | Instrument name |
<run name> | Upper and lowercase letters, integers 0-9, and underscores (_) | Run name as defined during run setup |
<flowcell ID> | Upper and lowercase letters and integers 0-9 | Flow cell ID from the barcode scan. If no barcode is present, the Run ID replaces the Flow cell ID. |
<lane> | 1 or 2 | Lane number |
<tile> | An integer | Tile number |
<x_pos> | A zero-padded integer | X-coordinate of the polony |
<y_pos> | A zero-padded integer | Y-coordinate of the polony |
<UMI> | A,T,C,G,N,+ | UMI sequence with a plus sign separating the Read 1 and Read 2 sequences, if applicable |
<read> | 1 or 2 | Read number |
<is filtered> | N | A legacy filtering value of N. The value exists only for backwards compatibility and does not change. |
<control number> | 0 | A legacy control number of 0. The value exists only for backwards compatibility and does not change. |
<indexsequence> | A value that depends on the indexing strategy indicated in the run manifest: - No indexing - The sample number - Single indexing - The observed index sequence - Dual indexing - The observed Index 1 sequence, a plus sign, and the observed Index 2 sequence |
Quality Scores
A Q-score indicates the confidence of a base call based on the Phred scale. A Phred quality score (Q) is logarithmically related to error rate (E): Q = -10log E. In a FASTQ file, an ASCII code represents the Q-score. Bases2Fastq encodes quality scores with a +33 offset (Phred33).
get table from noveed
Run Metrics
A run metrics file,RunStats.json, reports the following performance metrics in JSON file format. The metrics are specifiation the Bases2Fastq run.
| Metric | Value |
|---|---|
| AnalysisVersion | The current version of Bases2Fastq |
| AnalysisID | The unique,Bases2Fastq-generated identifier for the analysis |
| AssignedYield | The run yield based on assigned reads in gigabases |
| FileVersion | The current versionof the file format |
| FlowCellID | A flowcell identifier sourced from RunParameters.jsonor, if blank,the letter R followed bythe RunID value |
| I1IsReverseComplement | The observedorientationof the Index1 sequences relative to the orientation recordedin the run manifest |
| I2IsReverseComplement | The observed orientation of the Index 2 sequences relative to the orientation recorded in the run manifest |
| Lanes | A detailedlist of per-lanemetrics |
| MeanReadLength The average read length after adapter trimming | |
| NumPolonies | The totalnumber of polonies calculated for the run |
| NumPoloniesBeforeTrimming | The total number of polonies calculated for the run before adapter trimming |
| PercentAssignedReads | The percentage of reads assigned to a sample |
| PercentUnexpectedIndexPairs | The percentage of all polonies with Index1 and Index2 reads that matched different samples. |
| PercentMismatch | The percentage of polonie sassigned to a sample with mismatch |
| PercentQ30 | The percentageof ≥ Q30 Q-scores for the run, includingassignedand unassigned reads |
| PercentReadsTrimmed | The percentage of reads that Bases2Fastq trimmed |
| QualityScore10thPercentile | The 10th percentileof quality scores |
| QualityScore25thPercentile | The 25th percentileof quality scores |
| QualityScore50thPercentile | The 50th percentileof quality scores |
| QualityScore75thPercentile | The 75th percentileof quality scores |
| QualityScore90thPercentile | The 90th percentileof quality scores |
| QualityScoreHistogram | A per-base call Q-score distribution with integer resolution |
| QualityScoreMean | The averageQ-score of base calls for a sample |
| RunName | A text-based run identifiersourced from RunParameters.json |
| RunID | A universally uniqueidentifier (UUID)assigned to the run and sourced from RunParameters.json |
| Samples | A list of libraries the run sequenced |
| SampleStats | The per-sample metrics listed in the sample metrics files for the run |
| TotalYield | The total yield of all reads in gigabases |
| UnassignedSequences | A list of unassigned index sequences with a count for each unassigned sequence |
Sample Metrics
A sample metrics file reports the following sample-specific performance metrics in JSON file format. Bases2Fastq names the file per the convention{sample}_stats.json.
| Field | Value |
|---|---|
| AnalysisVersion | The current version of Bases2Fastq |
| BaseComposition | Counts for each A, C, G, T, and N base |
| ExternalID | An ExternalID specified in the run manifest, if applicable |
| FileVersion | The current version of the file format |
| MeanReadLength | The averageread length after adapter trimming |
| NumPolonies | The total numberof polonies assigned to the sample |
| NumPoloniesBeforeTrimming | The number of polonie sassigned to a sample before adapter trimming |
| Occurrences | Additional information per occurrence of the sample |
| PercentMismatch | The percentage of polonies assignedto the sample with mismatch |
| PercentQ30 | The percentage of ≥ Q30 Q-scores for the sample |
| PercentQ40 | The percentage of ≥ Q40 Q-scores for the sample |
| PercentReadsTrimmed | The percentage of reads that Bases2Fastq trimmed |
| PerReadGCCountHistogram | A list of counts: the valueat index i is the number of reads withi G/C calls |
| QualityScoreMean | The mean Q-score of base calls for the sample |
| RemovedAdapterLengthHistogram | A histogram showing the number of bases trimmed from an adapterin a given position |
| SampleID | A globally unique sample identifier |
| SampleName | The alphabetical sample identifier |
| SampleNumber | The numeric sample identifier |
| RunName | A text-based run identifier sourced from RunParameters.json |
| RunID | A UUID assigned to the run and sourced from RunParameters.json |
| Yield | The number of bases in the samplein gigabases |
Occurrences
Occurrences are a set of fields in a sample metrics file that allocate sample performance metrics by specific occurrences of a sample in the run. For example, if a sample appears in both lanes, Bases2Fastq lists an occurrence for each lane.
Each occurrence includes the identifiers Lane and ExpectedSequence and reports the following performance metrics.
| Occurrences | Field Value |
|---|---|
| BaseComposition | Counts for each A, C, G, T, and N base |
| CustomMetadata | Custom metadata specified in the run manifest, if applicable |
| MeanReadLength | The average read length after adapter trimming |
| NumPolonies | The total numberof polonies assigned to the sample |
| NumPoloniesBeforeTrimming | The number of polonies assignedto a sample before adapter trimming |
| PercentMismatch | The percentage of polonies assigned to the sample with mismatch |
| PercentQ30 | The percentage of ≥ Q30 Q-scores for the sample |
| PercentQ40 | The percentage of ≥ Q40 Q-scores for the sample |
| PercentReadsTrimmed | The percentage of reads that Bases2Fastq trimmed |
| PerReadGCCountHistogram | A list of counts: the value at index i is the numberof reads with i G/C calls |
| QualityScoreMean | The mean Q-score of base calls for the sample |
| R1Adapters | The Read 1 adapter sequences associated with the lane the occurrence belongs to |
| R2Adapters | The Read 2 adapter sequences associated with the lane the occurrence belongs to |
| RemovedAdapterLengthHistogram | A histogram showing the numberof bases trimmed from an adapter in a given position |
| Yield | The numberof bases in the sample in gigabase |
Troubleshooting
The following table provides resolutions to common problems that can occur during FASTQ file generation. If a problem persists, contact Element Technical Support.
| Problem | Resolution |
|---|---|
| Bases2Fastq fails to detect credentials attached to an AWS storage location with an EC2 instance | When using automaticrole detectionin AWS,makesure the regionenvironment variable is set credentials attached to an correctly in the EC2 instance: export AWS\_DEFAULT\_REGION=$aws\_region. |
| Indexing performance does not meet specifications | Make sure the run manifest includes the PhiX Control Library index sequences. Spiking in PhiX Control Library without recording the index sequences affects index assignment. |
| Indexing performance does not meet specifications | Review the index charts in the HTML QC report. The charts show the index assignment percentage rate, the number of polonies assigned to each index, and the most frequent unassigned indexes. Use this information to correct the run manifest or QC-fail the run. |
| Indexing performance does not meet specifications | Review the I1IsReverseComplement and I2IsReverseComplement metrics in RunStats.json. The metrics show the observed orientation of index sequences relative to the orientation recorded in the Index1 and Index2 columns of the run manifest. If a column contains inconsistent orientations, correct the run manifest accordingly. |
| The HTML QC report is missing from the output. | If the system is configured for static binary, make sure Python 3.6 or newer is installed with the necessary packages. Bases2Fastq always generates the report on Docker-configured systems but static binary requires Python 3.6 or newer |
| The HTML QC report is missing from the output. | If Python is correctly installed or the system is configured for Docker, review the error in info/QCReportErrors.txt for the cause. Use this information to generate the HTML QC report. |
| Flow cell ID is missing from the output. | Run Bases2Fastq with the flow cell ID argument to add a flow cell ID. For example:--flowcell-id "1234567890". |
| A corrected run manifest requires reprocessing the run. | Run Bases2Fastq with the QC-only argument to validate indexes on one tile in a corrected run manifest. For an example command, see Run QC-Only Mode on page 18. |