Cell assignment logic
After a run completes, Cells2Stats reads the in situ sequencing data for each cell, compares detected reads against the spacer library in your target cell assignment manifest, and assigns each cell to a dominant spacer. This page explains how that process works, what the default thresholds are, and how to interpret the results.
How cell assignment works
For each cell, the system:
- Detects reads at the target site using the cycles defined by the
TargetMask. - Matches each read against spacer sequences in the
[TARGET INFORMATION]section of your target manifest. - Calculates the confidence score per cell, a measure of the confidence of spacer assignment to the cell.
- Assigns the cell to the dominant spacer (
MostAbundantTarget) or leaves it unassigned based on whether the confidence score meets the minimum thresholds.
Read alignment
Reads are aligned to the spacer reference with bowtie in end-to-end, full-length, ungapped, strand-specific mode with an assigned mismatch threshold. Every alignment starts at read position 1, spans the entire read, has no indels or soft-clipping, and must match the supplied sequence in the forward orientation.
Read alignment parameters can be adjusted to tune read alignment. Depending on the direction of the change, adjustments can affect specificity or ambiguity risk in large libraries. Loosening tolerances increases ambiguity, while tightening them increases specificity at the cost of recovering fewer reads.
Read alignment parameters
| Parameter | Default | Adjustable? |
|---|---|---|
| Full-length alignment | Required | Yes, shorten Y cycles in TargetMask (for example, Y12N*) |
| Mismatch threshold | Assigned 1 mismatch per 16 spacer base pairs
| Yes, set per target overrides in the manifest with TargetMismatchThreshold, or override globally for all target sites with --target-mismatch-threshold when executing Cells2Stats. If both are set, the CLI argument takes precedence and is applied to all target sites. |
| Alignment start position | Base 1 | Yes, prefix TargetMask with N* cycles (for example, N4Y16) |
| Strand-specific alignment | Required | No, verify orientation in the manifest matches the 3' → 5' sequencing direction |
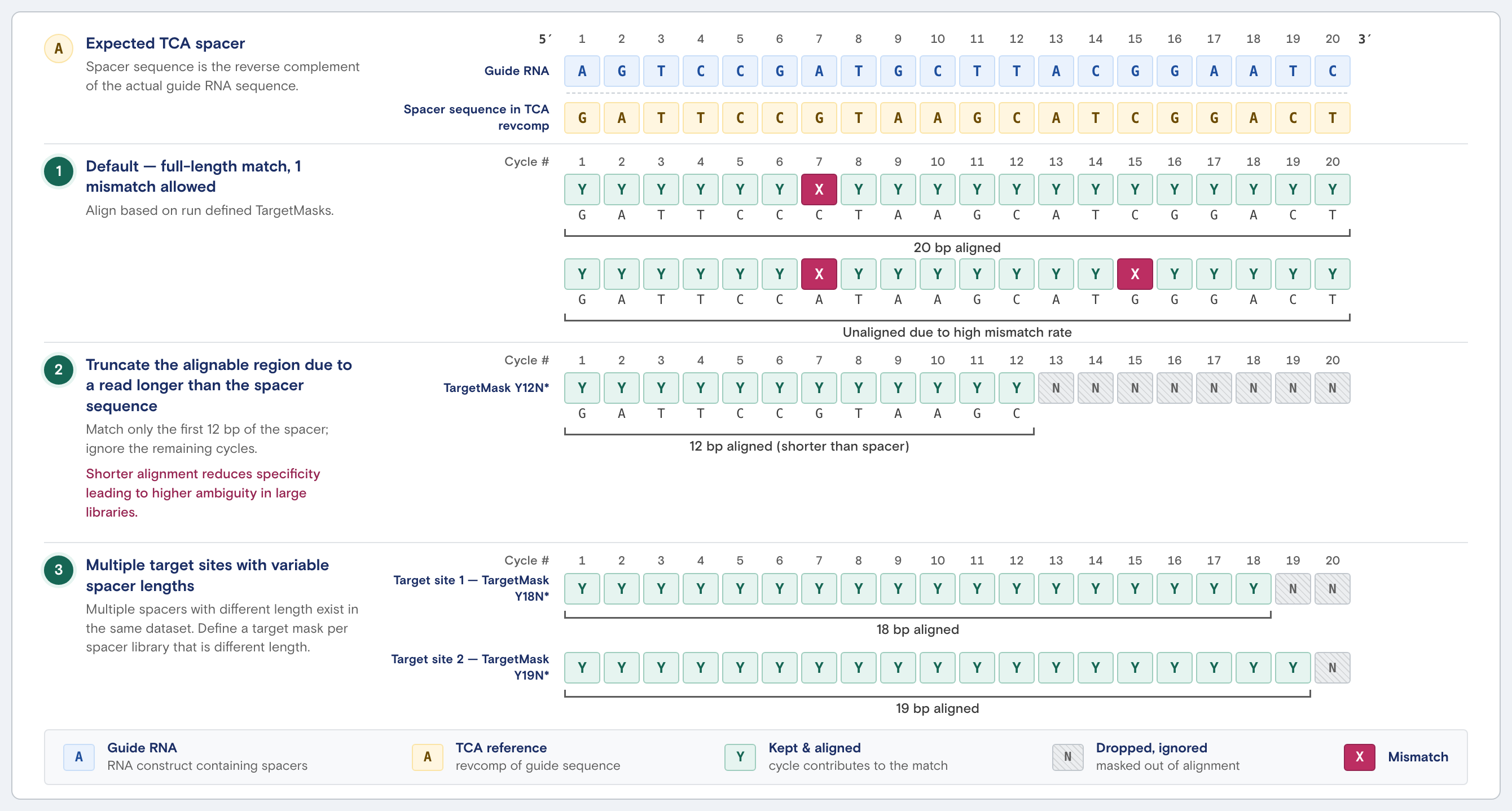
Default cell assignment parameters
Reads are aligned to sequences in the target cell assignment manifest. The sequencing output is the reverse complement of the actual guide RNA sequence. Thus, spacer sequences supplied in the TCA manifest must be provided in the same direction of sequencing. Each cell is assigned a confidence score.
Confidence score
The confidence score is calculated as: Confidence = MostAbundantTargetCount / (TotalCount + 1)
The +1 in the denominator (Laplace smoothing) penalizes cells with very little evidence. A cell with only 1 matching read scores 1 / (1 + 1) = 0.5, which does not exceed the threshold and is left unassigned. The single-read exclusion is therefore a consequence of the formula, not a separate rule.
Default threshold is greater than 0.5.
Pure cells vs. mixed cells
A key concept for interpreting results is the distinction between pure and mixed assigned cells.
| Category | Definition | RunStats.json field |
|---|---|---|
| Pure (assigned) | Cell has reads matching only one spacer. | PercentAssignedPureCells |
| Mixed (assigned) | Cell has reads matching multiple spacers, but one spacer dominates by read count and exceeds the confidence score threshold. | PercentAssignedMixedCells |
| Unassigned — mixed | Multiple spacers detected; no spacer exceeds the confidence score. | PercentUnassignedMixedCells |
| Unassigned — low count | No more than one spacer was matched and no spacer exceeds the confidence score threshold (0.5). | PercentUnassignedLowCountCells |
Both mixed-assigned and unassigned-mixed cells increase with higher multiplicity of infection (MOI). A high proportion of unassigned-mixed cells specifically signals that MOI may be too high to compromise reliable single-guide assignment, since no dominant spacer can be distinguished with confidence.
Pure vs mixed cell assignment The following example explains how for assigned reads a confidence score is mapped for various scenarios.
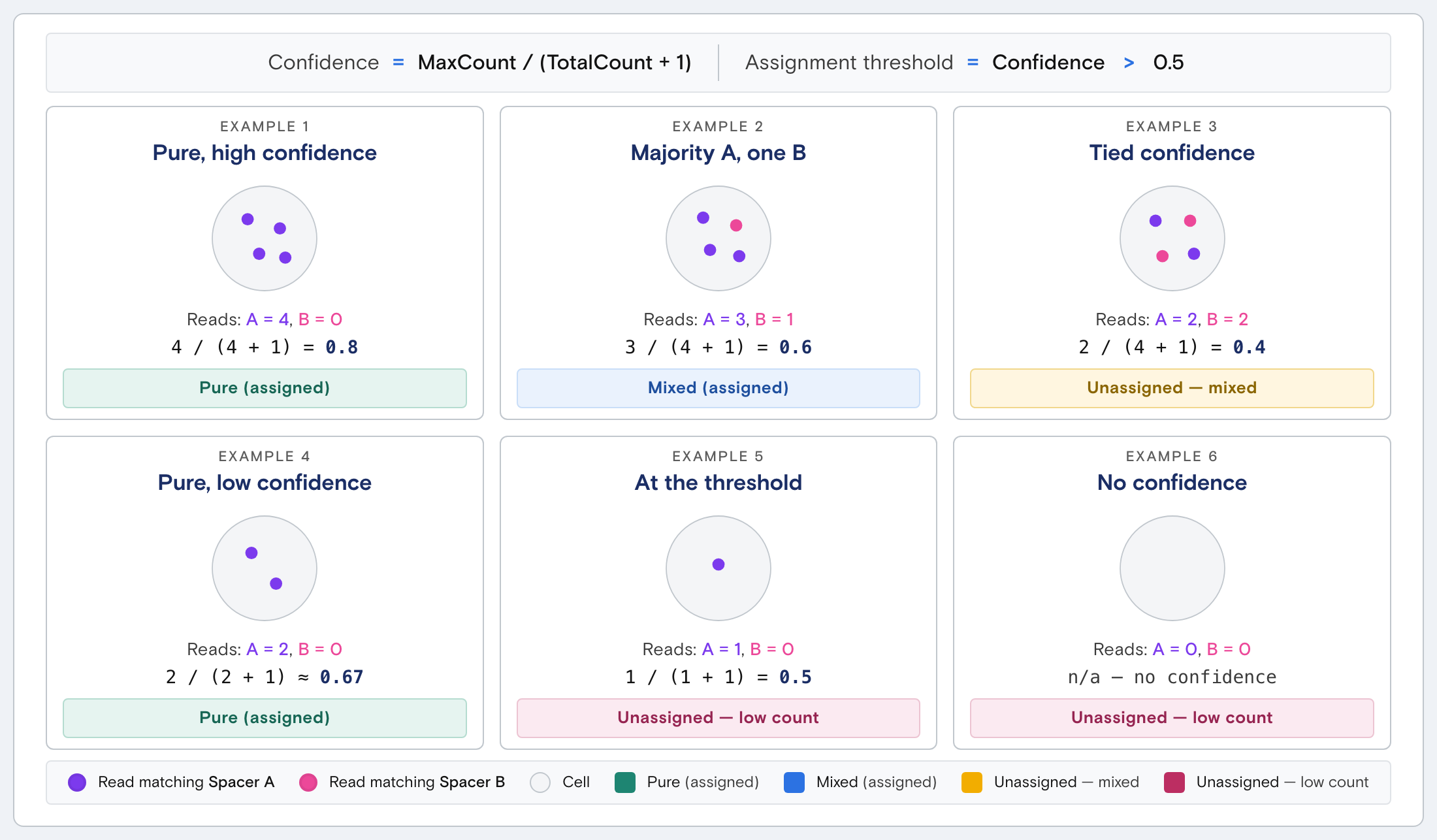
| Example | Confidence score | Outcome |
|---|---|---|
| 4 reads total , all match spacer A | 4 / 5 = 0.8 | Assigned to spacer A (pure) |
| 4 reads total, 3 reads match A, 1 matches B | 3 / 5 = 0.6 | Assigned to spacer A (mixed) |
| 4 reads total , 2 reads match A, 2 match B | 2 / 5 = 0.4 | Unassigned — mixed (ambiguous) |
| 4 reads total , 2 reads, both match A | 2 / 3 ≈ 0.67 | Assigned to spacer A (pure) — minimum reads to achieve assignment |
| 4 reads total , 1 read matches A, nothing else | 1 / 2 = 0.5 | Unassigned — does not exceed 0.5 |
| 4 reads total , 0 reads match | n/a | Undetermined — counted as low count |
Troubleshooting low assignment rates
Symptom (RunStats.json) | Likely cause | Resolution |
|---|---|---|
Nearly all cells unassigned; UnassignedSequences shows spacer-like reads | TargetMask misaligned OR spacer sequences in wrong orientation in target manifest | Check Panel.json for the mask applied; compare to spacer length and construct. Check sequence orientation in target manifest. |
| Manifest upload failed in Custom Designer | Sequence length / TargetMask mismatch | Ensure all sequences under each TargetSiteName are identical in length and match the Y cycle count. |
UnassignedSequences contains reverse complements of spacers | Spacer sequences in wrong orientation in target manifest | Reorient spacers such that they are the reverse complement of the actual guide RNA sequence. |
Known spacers appear in UnassignedSequences with high counts | Spacers missing from manifest | Add missing entries to [TARGET INFORMATION]. |
High PercentUnassignedLowCountCells; low MedianAbundantTargetCount | Low expression | Review RT primer binding efficiency; reach out to Element Technical Support for guidance. |
High PercentUnassignedMixedCells; high MeanUniqueTargetsPerCell | MOI too high | Optimize transduction conditions in future experiments. |
For a full reference of Cells2Stats output metrics and files, see Output files.